Zinc is important for tRNA recognition
[13].
Specificity determinants within tRNA
Ala that are important for recognition by AlaRS have been identified
[14, 15, 16, 17, 18, 19, 20]. A single nucleotide base pair in the acceptor helix of tRNA
Ala, G3-U70, is necessary and sufficient for aminoacylation of that tRNA with alanine
[21, 22, 23, 24, 25, 26, 27]. A mutation in
alaS which compensates for a mutant tRNA
Ala containing a G3-C70 base pair has been isolated
[28]. Discrimination of the G3-U70 base pair maps to a 76 amino acid region outside the catalytic center of AlaRS
[29]. The nucleotide at position 73 modulates the efficiency of the transfer step of aminoacylation
[30, 31, 32, 33].
Specificity determinants and residues within AlaRS that are important for catalytic activity have been investigated
[34, 35]. An N-terminal 461 amino acid fragment of the AlaS polypeptide was shown to complement a temperature-sensitive
alaS allele
in vivo; the C-terminal portion of the enzyme appears to be dispensable for catalytic activity, but is required for formation of the tetramer
[6, 36, 37, 38]. Mutagenesis of an N-terminal domain peptide reveals residues that increase catalytic activity of the fragment
[39]. A central region of AlaRS is essential for interaction with alanine-specific tRNA
[40]. Site-directed mutagenesis has identified the Arg69 residue within motif 2
[41, 42], the Cys665 residue
[43] and the Asp235 residue
[44] as important for catalysis. The Lys73 residue is important for recognition of tRNA
Ala [45, 46].
Single turnover kinetics experiments showed that a step prior to aminoacyl transfer is rate limiting
[47]. Kinetic parameters for binding of AlaRS to tmRNA have been determined
[48].
The central domain of AlaRS is the editing domain. Many aminoacyl-tRNA synthetases have been shown to have editing functions. AlaRS misactivates glycine and serine, but has a pre-transfer editing function, hydrolyzing the non-cognate amino acid before transfer to tRNA
Ala, and a post-transfer editing function that deacetylates mischarged tRNA
Ala [49, 50]. A series of crystal structures, together with kinetic analysis and mutagenesis, has allowed elucidation of the basis for recognition of alanine by AlaRS and provides a structural and evolutionary explanation for the interaction with serine
[51]. The covalently continuous two-domain structure of the tRNA is required to enable editing
[50]. The editing domain alone is able to specifically recognize mischarged tRNA
Ala, utilizing the same discriminator base pair within the tRNA as the aminoacylation domain
[52]. It deacylates misacylated Ser-tRNA
Ala with a k
cat/K
M of 6.6 x 10
5 M
-1 s
-1. The site-directed mutants Q584H, I667E, and C666A have decreased editing activity; interestingly, the C666A mutation is 1.7-fold more active on Ala-tRNA
Ala than on the mischarged Ser-tRNA
Ala [53]. A homologous free-standing protein with editing activity, AlaX, exists in other organisms; the protein family may have evolved from an ancestral form that was able to deacylate multiple tRNAs
[54]. |FRAME: CPLX0-3581| is able to edit Gly-tRNA
Ala more efficiently than AlaRS
[55].
The C-terminal domain (C-Ala) plays a role in activating the catalytic sites of the N-terminal domain
[56]. C-Ala represents the major tRNA binding module of AlaRS
[57]; this tRNA binding activity is required for efficient editing activity by the central editing domain
[52]. The C-Ala domain serves to bring the aminoacylation and editing domains of AlaRS together to bind to the acceptor stem of tRNA
Ala [57] and is important for dimerization of the enzyme
[58].
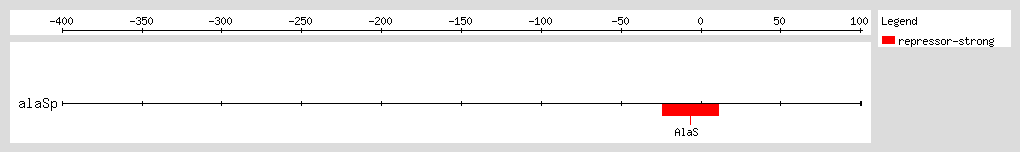
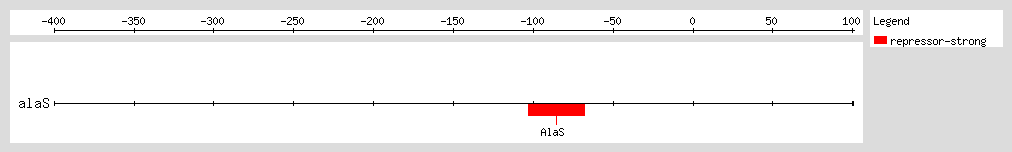
AlaRS represses transcription of the
alaS gene by binding to a region flanking the transcription start site, and thus autoregulates its own expression. The autoregulatory effect depends on the concentration of alanine, with higher concentrations leading to lower levels of
alaS transcription. At physiological levels of AlaRS, repression of
alaS transcription is solely mediated by alanine levels
[1].
The designed
alaS triple mutant T567F/S587W/C666F is unable to edit mischarged Ser-tRNA
Ala and Gly-tRNA
Ala. When combined with a
dtd deletion, the strain shows a synthetic growth defect and becomes sensitive to added glycine
[55]. The
alaS21 allele leads to increased resistance to novobiocin
[59]. Mutations in ribosomal proteins S5 and S20 partially suppress the temperature sensitive growth defect of an
alaS mutation
[60]. The mechanism of suppression is thought to be a reduction in the rate of polypeptide synthesis
[61].
Reviews:
[5, 62, 63, 64, 65, 66, 67, 68]